How Computer Scientists are Setting the Stage to End Tuberculosis
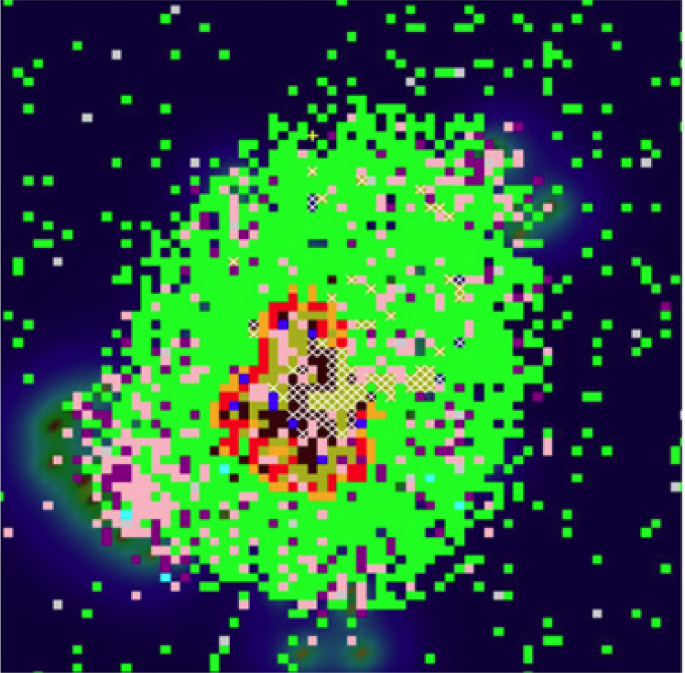 A model of a granuloma Source.
A model of a granuloma Source.
Today, March 24th, is World TB day, an annual commemoration of the day in 1882 when Robert Koch (a popular fellow on [the ASM] blog) announced his discovery that Mycobacterium tuberculosis (Mtb) is the bacterium responsible for tuberculosis (TB). Mtb is, according to the World Health Organization, “a globally established priority for which innovative new treatments are urgently needed.” That’s because nearly one third of the world population is infected with Mtb. There were 10.4 million new infections in 2015 and 1.8 million deaths due to TB worldwide. On a positive note, the TB infection rate has declined by an average of 1.5% per year since 2000, but 4-5% per year decline is needed to achieve benchmarks set by the “End TB Strategy.” Fittingly, the theme for the 2017 World TB Day is “Unite to End TB.”
](/project/setting-the-stage-to-end-tuberculosis/image_1_huc20e566429e7b53cf7da23c4c77a6f87_35553_00a3f11350505fa75a0ea12b293e9516.jpg)
{kind=link}
Collaborating to research global health threats is nothing new to scientific research and advances in research methodology, techniques and resources can take collaboration to new heights. This kind of innovation is sorely needed to study a pathogen as prolific and frustrating as Mtb. One of the conundrums facing TB research is the fact that Mtb grows incredibly slowly. Slow growth confounds both research and treatment since it can take days to weeks to grow a culture for study, a month or more to determine drug-resistance properties of a patient sample and requires months of antibiotic therapy once an infection is confirmed. At the University of Michigan, two labs have united to develop research strategies that bypass this limitation to provide new insights about TB. By using computer models, labs run by Denise Kirschner, Ph.D. and Jennifer Linderman, Ph.D., MSE are generating important insights into TB treatment, vaccines, and disease progression that might speed up the process of ending TB.
Known as in silico research, computer modeling compiles lab measurements of immune response or bacterial properties that occur during an infection to create programs that simulate potential outcomes. By altering the conditions of the modeled infections (like antibiotic treatments), in silico research can perform hundreds or thousands of complex experiments in moments, identifying new mechanisms for researchers at the bench to explore further. (Kirschner lab alum, Hayley Warsinske, Ph.D., does a much better job explaining the complexities of in silico models here.
Introducing the tuberculosis cast
Modeling an event as complex as a Mtb infection and the corresponding immune response requires a nuanced understanding of the events that occur. TB infection begins with inhalation of Mtb bacilli from an infected individual by an uninfected person. In the lungs, early responders of the innate immune response known as macrophages engulf the bacteria, attempting to destroy the invaders. However, Mtb is quite durable, employing many strategies to avoid killing by macrophages that allow survival and continued growth of the bacilli, both within the lungs and even the hostile environment of macrophages. The death of the macrophages releases messenger molecules called cytokines that recruit more innate immune cells such as monocytes (immature macrophages) to continue battling Mtb.
At the same time, scout cells such as dendritic cells, collect Mtb samples and carry them to the nearest lymph node, a command center for the adaptive immune response. The dendritic cells display degraded Mtb proteins on their cell surfaces, looking for T cells that recognize the peptides. When a T cell match is found, cytokines instruct the T cell as to the type of immune response to orchestrate (inflammatory vs anti-inflammatory). The types of cytokines released by the dendritic cells and the proportion of different cytokines to the others are key in directing the T cell response. The newly activated anti-Mtb T cells then migrate to the site of infection. This process takes about 2 months and results in a Mtb-specific immune response.
, Fig. 3c)](/project/setting-the-stage-to-end-tuberculosis/image_2_huda7038dc17ea33dbfaf8897787b2eaee_1889904_29875facbb817a7d0e064a50fa33ce17.png)
When the Mtb-specific T cells respond, they produce more cytokines that stimulate (activate) the macrophages, enhancing their ability to kill intracellular Mtb. This leads to the formation of a granuloma, a cocoon of macrophages (active and inactive) around surviving bacteria that prevents them from spreading any further. In some cases, the bacteria are eventually cleared from the granulomas (sterilized). In others, latent Mtb, the bacteria survive within the granulomas but the infection remains asymptomatic. Such infections can later be reactivated, a process where Mtb escapes the granuloma causing further damage to the lungs and re-initiating the immune response.
Setting the model stage
According to Joey Cicchese, a graduate student in the Linderman lab, the crux of in silico modeling is “translating the mechanisms that govern biological processes into computer code.” To translate these mechanisms, or events, into code, Kirschner and Linderman need numbers: how many macrophages are present and how many are active, how many bacteria are alive, what cytokines are produced and to what extent, how many T cells are recruited, how many granulomas are formed, how many are sterilized, how many latent granulomas are reactivated, etc. And all of this from different time points during an infection. For this, they rely on data from collaborators studying TB in animal models such as non-human primates (JoAnne Flynn, Ph.D.) and rabbits (Véronique Dartois, Ph.D.).
“The code is then used to run simulations that mimic the biological process.” –Cicchese.
The vast number of events, molecules, and cells must all be taken into account, so the modeling occurs at different levels of magnification: the cellular level (between macrophage and bacilli or dendric cell and T cell), the tissue level (granulomas in the lung, lymph nodes, T cell migration or drug dynamics in the blood), the body level (a single virtual patient), and the population level (many virtual patients). Results from more magnified models can, in turn, be scaled up to populate the next lower magnification model.
The real power behind in silico modeling, Cicchese notes, is that “during our granuloma simulations, we can keep track of bacteria and each immune cell counts at any time throughout the simulation.” He goes on to add that “because in silico modeling is typically faster than most wet lab experiments (e.g., from Flynn and Dartois), it can be a way to guide experimentalists to studies that are more likely to succeed or produce interesting results.”
Break a leg – identifying new hypotheses
Just in the last couple of years, the group has published at least three intriguing models or hypotheses for experimentalists to follow up on. TB treatment regimens last months and require the use of multiple antibiotics to prevent drug resistance, problems that confound research into the efficacy of new therapeutics. A 2015 study in BMC Systems Biology published the combination of drug pharmokinetics modeled in the blood with tissue pharmokinetics and granuloma development models. This combined model system provides the ability to look at drug dynamics at many different levels, which could predict odds of an infection developing drug resistance under different dosages and frequencies. Additionally, it could predict the efficacy of immune modulation as a treatment strategy and predict the likelihood of treatment regimens in clinical trials. If accurate, these models would improve the success of conducted trials saving time, money, and lives.
Another multi-level model published in PLoS Computational Biology identified biomarkers capable of predicting disease outcomes. There is a spectrum of outcomes between fully active TB infection and latent TB. This spectrum is influenced by many factors including the immune response, the bacterial strain, and treatment strategy. Being able to predict disease outcome in a lengthy disease like TB would allow clinicians earlier opportunities to alter treatment and improve prognosis. Previous attempts to identify biomarkers had been unsuccessful, in part because the best biomarkers should be easily accessed (e.g., via blood draw) but the cytokines or cell types present in the blood are not necessarily indicative of what’s occurring in the lungs. Using data derived from non-human primates, their model correlated Mtb-specific T cells with positive disease outcomes. The greater the proportion of Mtb-specific T cells, the more likely Mtb was to be contained. This gives experimental researchers a direction (identifying Mtb-specific T cells) to pursue in their search for a reliable biomarker.
Mtb-specific T cells are essential to effectively containing a TB infection and if activated by vaccination, the infection could be avoided altogether. However, eliciting a strong T cell response through vaccination is tricky and only one vaccine, BCG, is currently used to prevent TB. Administered in infancy, the vaccine largely loses efficacy by the time adolescence is reached. A granuloma model reported in Frontiers in Microbiology by Linderman and Kirschner tests the efficacy of various vaccine types, routes, and doses. While they caution that a larger system model needs to be developed to better understand what happens at the body and population level, this model would be valuable for virtual trials of new TB vaccines and predict vaccine components that will generate the desired response.
Collectively, in silico research offers experimental researchers a way to conduct dress rehearsals of expensive and complex hypotheses. But as Cicchese pointed out “even though in silico models produce more data, the simulated data are only as good as the experimental data.” Collaborations between computer scientists, wet lab researchers and clinicians are essential to making these systems work to improve TB research. The work pioneered by Lindermann, Kirschner, Flynn, and Dartois are just one example of how we can unite to end TB.
*Republished from ASM.org with permission
Ada K. Hagan, Ph.D.
Owner, Lead Consultant
I am a microbiologist with a passion for making science accessible. I hope to use my background in communications and higher education to help make scientific concepts more easily understood and make the academy more inclusive to future scientists from all backgrounds.